Life Sciences Workflow 2
Course objectives
The main objective of this course is to demonstrate how a bioinformatics workflow can be seamlessly run using TACC resources from raw input to biological interpretation. There are many different types of workflows you can run using the systems we have available here at TACC. In particular, transcriptome analysis will be demoed as it is a familiar concept among wide range of disciplines.
- Understand Transcriptome analysis
- Run through a RNA-seq data analysis pipeline using Stampede
1. Transcriptome analysis

Transcriptome is simply defined as all of RNA molecules in a single cell.
- Represents expression levels of all genes for a single condition of cell for a specie at given time.
- Also Rereferred as expression profiling.
- Unlike Genome analysis can show differences in the exact same cell that are in different external environment.
Only a decade ago, the study of gene expression was limited to human genetics for medical purposes or model organisms such as mouse, fruit fly and nematodes. Then, microarrays and serial analyses of gene expression were the only available tools for examining transcriptome. With the recent advances of next-generation sequencing technologies, the cost effectiveness of sequencing and maturation of analytical tools, the transcriptome analysis has become more a realistic option for genetic nonmodel organisms, even for individual laboratories.

a. Examples from high-impact journal



Transcriptome studies often give high-impact results as it describes status of cells in different conditions in context of expression levels. Here are some examples of these in Science, Nature and Cell (Impact Factor 34.661, 38.138, 28.710 respectively)
b. Scientific background
The pipeline uses Next-generation sequencing RNA-seq data to as a raw input. These are prepared by (experimental workflow).
https://www.nature.com/article-assets/npg/nrg/journal/v12/n10/images/nrg3068-f1.jpg
{kind=link}
2. Transcriptome analysis workflow using TopHat suite
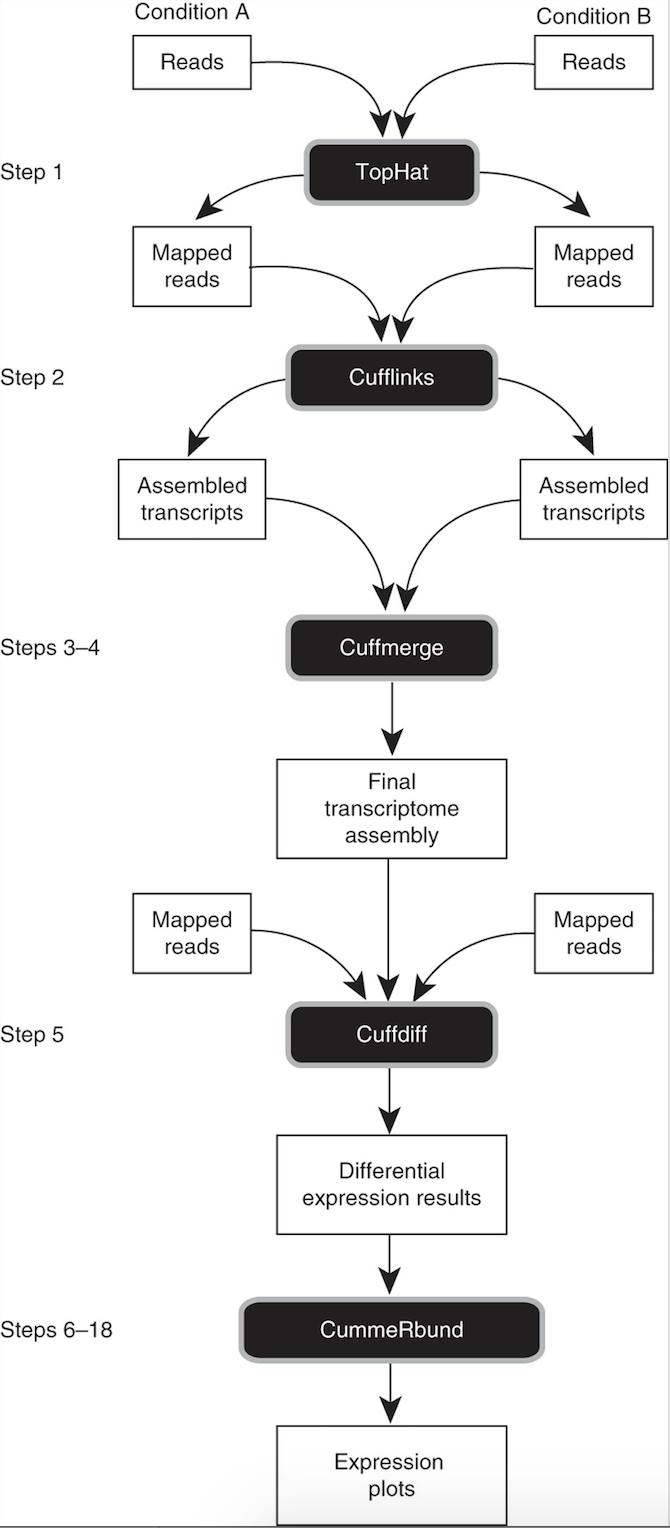
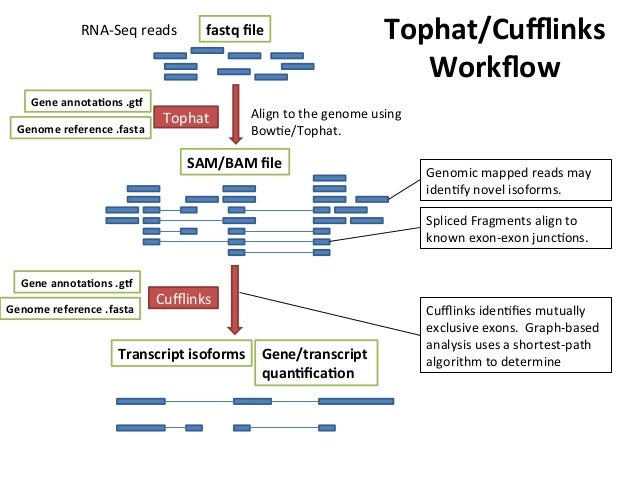
3. Where to find required test-dataset
Sequence Read Archive
https://www.ncbi.nlm.nih.gov/sra/SRX2771645 https://www.ncbi.nlm.nih.gov/sra/SRX2771647
Genome index and annotation https://ccb.jhu.edu/software/tophat/igenomes.shtml
- Hands-on: Try downloading and extracting genome index and annotation files to a directory
wget ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/Arabidopsis_thaliana/Ensembl/TAIR10/Arabidopsis_thaliana_Ensembl_TAIR10.tar.gz
tar -zxvf Arabidopsis_thaliana_Ensembl_TAIR10.tar.gz
4. Tools used in this session
SRAtoolKit
Tuxedo suite: includes: TopHat, Cufflinks, Cuffmerge, Cuffdiff (cite) https://www.nature.com/article-assets/npg/nprot/journal/v7/n3/images_article/nprot.2012.016-F1.jpg
{kind=link}
Samtools
5. Benchmarking analysis (TopHat)
What is the advantage of using TACC vs. workstation on transcriptome analysis? Testing number of threads and CPU time for an alignment to complete
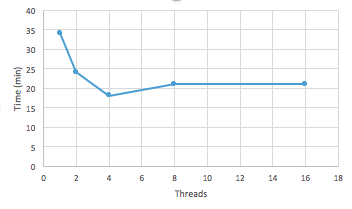
6. Information on test dataset (Scientific background)
The two sequencing files are of a plant model organism, Arabidopsis Thaliana.
- A member of the Brassica family
- A favorite research subject for plant molecular geneticists
- Due to its remarkably small genome size (about 100,000 kB, with less than 25% repetitive sequences)
- And ease of culture in the lab
- Short stature and life cycle
- Ameanable to genetic mapping studies and other techniques (such as transformation and gene cloning).

- Col-0 strain, which is columbia seed.
- A widely used wild type seed, selected for its high fertility, vigor, and responsiveness to changes in photoperiod.
-
Contains no visible genetic markers.
- Second is of jazQ mutant, which has enhanced JA-regulated defense against insect herbivory without an associated reduction in leaf growth.
- Is a JA signalling mutant (jazQ), in which the removal of multiple JAZ repressors causes hyperactivation of JA responses
- As a consequence, jazQ plants exhibit both enhanced resistance to insect herbivory and diminished growth of leaves and roots.
First hands on:
- Convert SRA file to FASTQ
module load sratoolkit scratch_cache prefetch SRR5488800 ; fastq-dump SRR5488800 prefetch SRR5488802 ; fastq-dump SRR5488802cp /scratch/02114/wonaya/SSI/SRR5488800.fastq . ; cp /scratch/02114/wonaya/SSI/SRR5488802.fastq .
(2 minutes)
7. TopHat and Cufflink demo/hands-on
module load perl bowtie tophat
module load boost on ls5
What happens if you do just module load tophat without prerequisite modules?
TopHat run: Aligning sequences on arabidopsis genome guided with gene annotations
Remember Do NOT run long processes (more than 5 minutes) on compute node
tophat2 -p 4 -G Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -o test_1 --no-novel-juncs Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome SRR5488800.fastq
tophat2 -p 4 -G Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -o test_2 --no-novel-juncs Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome SRR5488802.fastq
cp -r /scratch/02114/wonaya/SSI/test_1/ . ; cp -r /scratch/02114/wonaya/SSI/test_2/ .
(15*2 minutes)
Cufflinks:
cufflinks -o wt_cuff -G Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf test_1/accepted_hits.bam
cufflinks -o sa_cuff -G Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf test_2/accepted_hits.bam
cp -r /scratch/02114/wonaya/SSI/wt_cuff/ . ; cp -r /scratch/02114/wonaya/SSI/sa_cuff/ .
(7*2 minutes)
Cuffmerge:
cuffmerge -g Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf -s Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/WholeGenomeFasta/genome.fa -p 16 cuffmerge.txt
in which the content of cuffmerge.txt should be (using text editor such as vim):
wt_cuff/transcripts.gtf
sa_cuff/transcripts.gtf
cp /scratch/02114/wonaya/SSI/cuffmerge.txt .
(2 minutes)
Cuffdiff:
cuffdiff -L WT,SA -p 16 merged_asm/merged.gtf test_1/accepted_hits.bam test_2/accepted_hits.bam
cp -r /scratch/02114/wonaya/SSI/merged_asm .
(11 minutes)
8. Alignment statistics
cat test_1/align_summary.txt
How efficient was the alignment? TopHat reports alignment statistics internally, but other tools may not. In these cases, use samtools flagstat
9. Functional analysis with BARtools
So with this data, what can you deduce?
http://bar.utoronto.ca/ntools/cgi-bin/ntools_classification_superviewer.cgi
Using the dataset you got from running cuffdiff, you should be able to extract top 100 genes with highest log-fold changes in expression. Save the names of these genes as a separate text file, and copy the names of genes to the tool above. What functional enrichment do you see in mutant strain?
sort -nrk 10 gene_exp.diff > gene_exp.diff.sort
head -100 gene_exp.diff.sort > gene_exp.diff.sort.top100
cut -f 3 gene_exp.diff.sort.top100 > gene_exp.diff.sort.top100.names
or Can you pipe this into one process?
sort -nrk 10 gene_exp.diff | head -100 - | cut -f 3 - > gene_exp.diff.sort.nrk.top100.names
Results:
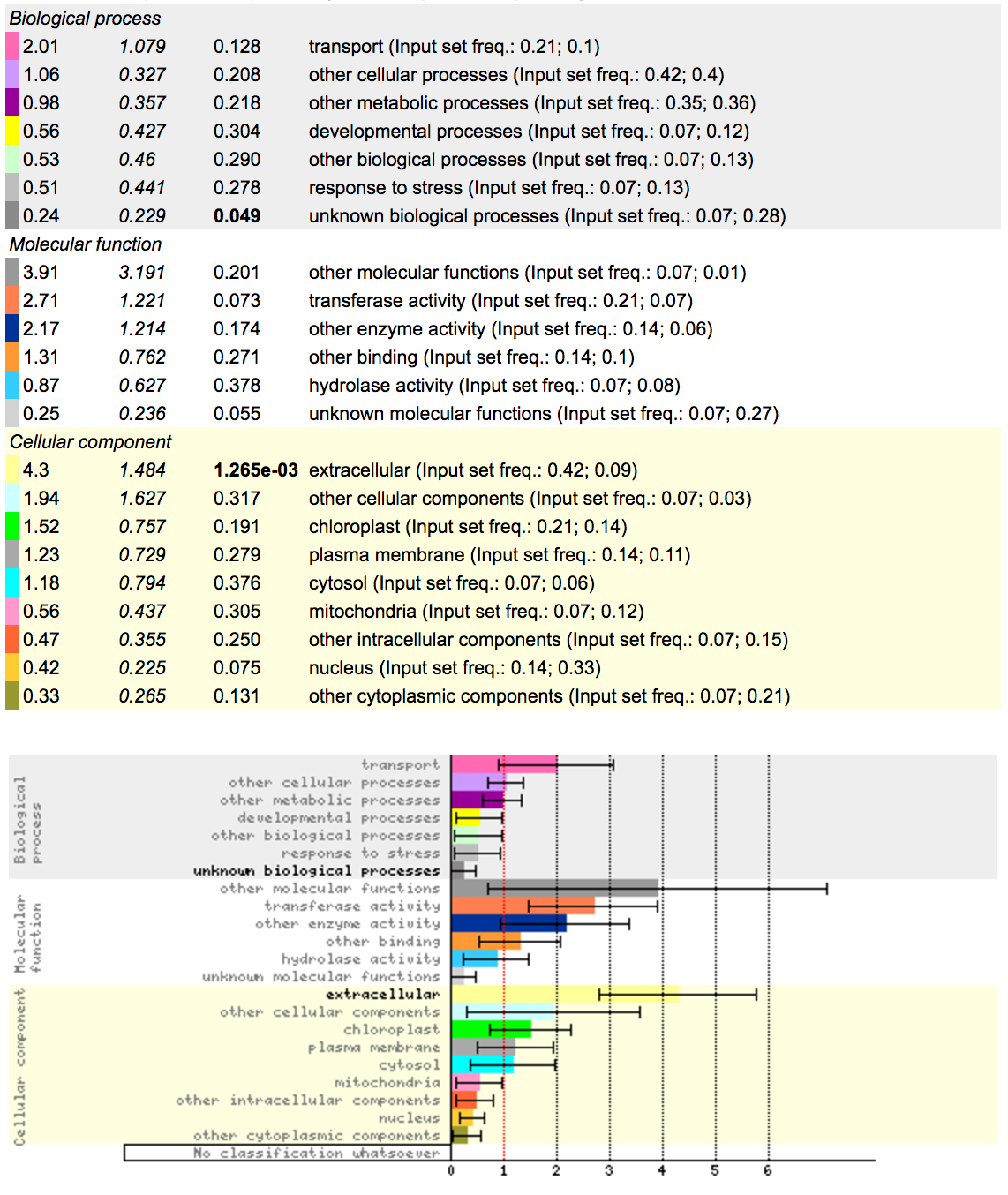
Removal of multiple JAZ genes in this jaz quintuple (jazQ) mutant led to constitutive activation of JA responses, causing hypersensitivity to exogenous JA treatment, upregulation of defense-related genes, increased production of secondary metabolites and higher resistance to insect herbivory attack Campos et al. Nat. Comms. 2016 https://www.ncbi.nlm.nih.gov/pubmed/27573094
Congratulations, You can now do Transcriptome analysis!
Hands-on
- Try submitting to TACC system using a job script for tophat to cuffdiff workflow
- Explore different RNAseq dataset
https://www.ncbi.nlm.nih.gov/sra/SRX2834995 https://www.ncbi.nlm.nih.gov/sra/SRX2834994